
domingo, 2 de marzo de 2014
SINDROME SAVANT
Se conoce como síndrome del savant o savantismo a un conjunto de síntomas cognitivos anómalos. Estos individuos son denominados savants, normalmente traducido como "sabios", siendo la traducción literal "sabedores" (sapientes). Otros investigadores indican que los rasgos y las habilidades autísticos del savant pueden estar ligados
Arte : se caracterizan por ser grandes intérpretes musicales, especialmente al piano, pintores y escultores. Suelen tener habilidades innatas para comprender e interpretar la música.
Cálculo de fechas: algunos savant pueden memorizar calendarios enteros y recordar datos referentes a cada uno de esos días.
Cálculo matemático: capacidad para la realización de complejos cálculos matemáticos mentalmente de forma instantánea y con gran precisión, como por ejemplo el cálculo de números primos o la realización de divisiones con 100 decimales mentalmente.
Habilidades mecánicas y espaciales: capacidad para medir distancias casi exactas sin la ayuda de instrumentos, construcción de detalladas maquetas, memorización de mapas y direcciones...
Existen además otra serie de habilidades, más inusuales y en general más particulares del individuo, como facilidad para el aprendizaje de múltiples idiomas, fuerte agudización de los sentidos, perfecta apreciación del paso del tiempo sin necesidad de relojes.
PORFIRIA
 Se conoce como ‘enfermedad de los vampiros’ a la porfiria, un extraño trastorno que normalmente se transmite por vía genética y que, entre otros síntomas, provoca ampollas en el cuerpo de quienes la padecen cuando existe exposición a la luz solar. Es esta particularidad precisamente la que inspira su vampírico sobrenombre.
Se conoce como ‘enfermedad de los vampiros’ a la porfiria, un extraño trastorno que normalmente se transmite por vía genética y que, entre otros síntomas, provoca ampollas en el cuerpo de quienes la padecen cuando existe exposición a la luz solar. Es esta particularidad precisamente la que inspira su vampírico sobrenombre.Más concretamente, las porfirias son un conjunto de enfermedades derivadas de una producción anómala de la hemoglobina, una proteína de la sangre. La deficiencia se produce en un grupo prostético llamado hemo, cuya biosíntesis es deficiente por problemas en las enzimas.

PICA
La Pica
Las personas que sufren de la condición médica conocida como Pica sienten un extraño deseo por comer objetos y sustancias que no son alimentos, que son peligrosos para el organismo y que aportan bajísimos niveles nutritivos al cuerpo. Comúnmente, se trata de objetos como clavos, metal, vidrio, madera, papel, piedras, etc. El extraño desorden se cree que se presenta a nivel físico como producto de bajos niveles de minerales en el cuerpo, mientras que a nivel psicológico, se presenta tras la exposición a largos períodos de estrés.Existen distintos tipos de Pica, en algunos casos ocurre con un fin cultural, siendo una suerte de ritual que a todos nos parece un tabú. Aún así, es una enfermedad, un trastorno físico y psicológico que muchas veces tiene consecuencias mortales. La intervención quirúrgica es un factor común en estos casos debido a obstrucciones intestinales y problemas estomacales.

ELEFANTIASIS
La elefantiasis es un síndrome caracterizado por el aumento enorme de algunas partes del cuerpo, especialmente de las extremidades inferiores y de los órganos genitales externos.
Puede producirse por diversas enfermedades inflamatorias persistentes, producida por diversas espiroquetas, treponemas y parásitos, muy especialmente en los países cálidos del grupo de la filaria, con más de 250 millones de casos registrados hasta 2007.Esta enfermedad se debe a la obstrucción de vasos linfáticos, y es por ello que se dan como resultado inflamaciones severas y de parásitos sanguíneos como las filarias (parásitos habitantes de los vasos linfáticos, de cavidades corporales, corazón y otros lugares del cuerpo dependiendo de la especie). También causa malformación de huesos, la cual va deformando el cuerpo hasta donde la deformación lo permita.


Puede producirse por diversas enfermedades inflamatorias persistentes, producida por diversas espiroquetas, treponemas y parásitos, muy especialmente en los países cálidos del grupo de la filaria, con más de 250 millones de casos registrados hasta 2007.Esta enfermedad se debe a la obstrucción de vasos linfáticos, y es por ello que se dan como resultado inflamaciones severas y de parásitos sanguíneos como las filarias (parásitos habitantes de los vasos linfáticos, de cavidades corporales, corazón y otros lugares del cuerpo dependiendo de la especie). También causa malformación de huesos, la cual va deformando el cuerpo hasta donde la deformación lo permita.
EPIDERMODISLAPSIA VERRUCIFORME
Epidermodisplasia verruciforme (también llamado displasia de Lewandowsky-Lutz epidermodisplasia verruciforme de Lutz-Lewandowsky) es una rara dominancia genética del trastorno de la piel hereditaria asociada a un alto riesgo de carcinoma. Se caracteriza por la susceptibilidad anormal al virus del papiloma humano (VPH).El resultado no controlado de infecciones por VPH trae como consecuencia el desarollo de escamas máculas y pápulas, sobre todo en manos y pies. Es típicamente asociados a tipos de VPH 5 y 8, que se encuentran cerca del 80% de la población normal, como las infecciones asintomáticas aunque otros tipos también pueden contribuir.
La condición suele comenzar entre las edades de 1 a 20 años , pero en ocasiones se puede presentar en personas de mediana edad. El nombre se debe a los dermatólogos que documentaron por primera vez un caso, Félix Lewandowsky (1879-1921) y Wilhelm Lutz (1888-1958).A los 35 años, Dede Koswara notó que de sus manos y pies surgían unos tejidos similares a las raíces de los árboles. Tras varias pruebas, los médicos detectado que el joven indonesio, sufría una condición extraña llamada epidermodisplasia verruciforme. Esta enfermedad es de origen genético.

La condición suele comenzar entre las edades de 1 a 20 años , pero en ocasiones se puede presentar en personas de mediana edad. El nombre se debe a los dermatólogos que documentaron por primera vez un caso, Félix Lewandowsky (1879-1921) y Wilhelm Lutz (1888-1958).A los 35 años, Dede Koswara notó que de sus manos y pies surgían unos tejidos similares a las raíces de los árboles. Tras varias pruebas, los médicos detectado que el joven indonesio, sufría una condición extraña llamada epidermodisplasia verruciforme. Esta enfermedad es de origen genético.
FETUS IN FETU
El fetus in fetus, gemelo parásito o mellizo parásito, es una formación humanoide creada por un accidente en cierto punto de la formación del cigoto antes de la formación del embrión.
Ocurre en 1 de 500,000 nacimientos, y se define como un humanoide que no tiene órganos internos funcionales. Al extraerse, está envuelto en lo que parece un huevo de gallina, pero mucho más grande y extremadamente duro.
El parásito, cuando es extraído y expuesto, tiene un color blanco. Una vez que se extrae, muere, ya que es un tejido totalmente dependiente de su hospedador. Es tejido humano, pero no se le puede considerar un ser humano. Es un caso raro de gemelos siameses.
Es un potencial riesgo para su hospedador, debido a que se alimenta de este, representando para el hospedador, una carga semiviva sin función en el organismo. No tiene cerebro, sino cráneo vacío. La cavidad torácica no existe, pues viene siendo, junto con la cavidad abdominal, carne maciza, y puede pesar hasta un kilogramo, pero sí tiene algunas características variables, como manos y hasta piernas. Es considerado un tumor.

Ocurre en 1 de 500,000 nacimientos, y se define como un humanoide que no tiene órganos internos funcionales. Al extraerse, está envuelto en lo que parece un huevo de gallina, pero mucho más grande y extremadamente duro.
El parásito, cuando es extraído y expuesto, tiene un color blanco. Una vez que se extrae, muere, ya que es un tejido totalmente dependiente de su hospedador. Es tejido humano, pero no se le puede considerar un ser humano. Es un caso raro de gemelos siameses.
Es un potencial riesgo para su hospedador, debido a que se alimenta de este, representando para el hospedador, una carga semiviva sin función en el organismo. No tiene cerebro, sino cráneo vacío. La cavidad torácica no existe, pues viene siendo, junto con la cavidad abdominal, carne maciza, y puede pesar hasta un kilogramo, pero sí tiene algunas características variables, como manos y hasta piernas. Es considerado un tumor.
Progeria de Hutchinson-Gilford
La progeria de Hutchinson-Gilford es un síndrome extremadamente raro caracterizado por un envejecimiento prematuro de inicio postnatal. La progeria puede afectar diferentes órganos y tejidos: hueso, músculos, piel, tejido subcutáneo y vasos. Desarrollan baja estatura, cráneo de gran tamaño, alopecia, piel seca y arrugada, ausencia de grasa subcutánea, rigidez articular. Al no existir cura ni tratamiento, las personas que lo padecen, no exceden los 13 o 15 años de vida.

Esclerosis Lateral Amiotrófica
La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa que causa una pérdida progresiva de las neuronas motoras. La prevalencia de la enfermedad es de 5 a 9 por cada 100.000 sujetos. La edad de aparición de la enfermedad varía ampliamente, pero el pico de incidencia se sitúa entre los 40 y los 60 años. En aproximadamente dos terceras partes de los pacientes de ELA se empieza con debilidad y deterioro muscular de miembros asimétrica. La enfermedad es implacablemente progresiva, con discapacidad y minusvalía en aumento, conduciendo generalmente a la muerte como consecuencia de insuficiencia respiratoria en un plazo de 3 a 5 años aproximadamente.
Aunque los síntomas tempranos varían de un sujeto a otro, todos los pacientes suelen mostrar los siguientes trastornos: se les caen los objetos, tropiezan, sienten una fatiga inusual en brazos o piernas, muestran dificultad para hablar y sufren calambres musculares y tics nerviosos.
SINDROME X FRAGIL
El síndrome X frágil es una enfermedad genética rara, debida a un defecto hereditario en el cromosoma X. Es la causa conocida más frecuente de retraso mental hereditario y la segunda cromosopatía después del síndrome de Down. Se estima que la frecuencia en España es de 1 por cada 4.000 varones en la población general, una portadora por cada 800 y un portador por cada 5.000 nacidos vivos. Clínicamente cursa con retraso mental de grado variable, aunque suele ser leve con dificultades en el aprendizaje, falta de atención, hiperactividad, con ansiedad y humor inestable o comportamientos autistas.
el Síndrome del Cromosoma X frágil no tiene tratamiento médico curativo, pero si tratamiento paliativo de algunos de sus síntomas.
El tratamiento ha de seguir dos vías esenciales, por un lado un tratamiento médico y por otro un tratamiento educativo.
El tratamiento médico debe ser impartido por especialistas y en general deben ir encaminados a mejorar determinados problemas
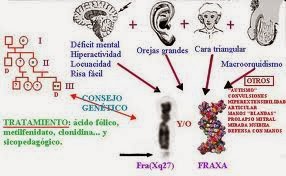
el Síndrome del Cromosoma X frágil no tiene tratamiento médico curativo, pero si tratamiento paliativo de algunos de sus síntomas.
El tratamiento ha de seguir dos vías esenciales, por un lado un tratamiento médico y por otro un tratamiento educativo.
El tratamiento médico debe ser impartido por especialistas y en general deben ir encaminados a mejorar determinados problemas
Síndrome de Apert
El síndrome de Apert es un tipo de acrocefalosindactilia, un trastorno congénito caracterizados por deformaciones en el cráneo, cara, manos y pies. Se suele clasificar como un síndrome del arco branquial, con afectación del primer arco branquial; que en los humanos es precursor del maxilar y mandíbula. Las perturbaciones en el desarrollo de los arcos branquiales en el desarrollo fetal provocan efectos duraderos y generalizados
En base a la evidencia empírica la acrocefalosindactilia podría tratarse de un desorden autosómico dominante. Niños y niñas se encuentran igualmente afectados; sin embargo queda por investigar la causa exacta. Sin embargo, casi todos los casos son esporádicos, implicando mutaciones de novo o algún tipo de daño ambiental sobre el genoma. La descendencia de un padre con el síndrome de Apert tiene una probabilidad del 50% de heredar la enfermedad. En 1995, A.O.M. Wilkie publicó un documento que muestra evidencia de que acrocefalosindactilia es causada por un defecto en el gen del receptor del factor de crecimiento de fibroblastos 2, ubicado en el cromosoma 10.


En base a la evidencia empírica la acrocefalosindactilia podría tratarse de un desorden autosómico dominante. Niños y niñas se encuentran igualmente afectados; sin embargo queda por investigar la causa exacta. Sin embargo, casi todos los casos son esporádicos, implicando mutaciones de novo o algún tipo de daño ambiental sobre el genoma. La descendencia de un padre con el síndrome de Apert tiene una probabilidad del 50% de heredar la enfermedad. En 1995, A.O.M. Wilkie publicó un documento que muestra evidencia de que acrocefalosindactilia es causada por un defecto en el gen del receptor del factor de crecimiento de fibroblastos 2, ubicado en el cromosoma 10.
Acidemia propiónica
La acidemia propiónica es una enfermedad hereditaria ocasionada por un déficit de la enzima propionil CoA carboxilasa. Esta enzima se encuentra en las mitocondrias de la célula y sirve para catalizar la transformación de propionil CoA a metilmanolil CoA, este paso metabólico forma parte de la degradación de los aminoácidos valina, metionina, treonina e isoleucina, interviene también en la metabolización de aquellos ácidos grasos que tienen un número impar de átomos de carbono. La deficiencia de la actividad enzimática ocasiona una concentración elevado de propionato en orina y sangre.
Este trastono se incluye dentro del grupo de enfemedades llamadas errores congénitos del metabolismo y su frecuencia es variable según la región, en Estados Unidos se presenta un caso por cada 35000 nacimientos, mientras que en Arabia Saudi afecta a 1 de cada 3000 nacidos.
Se hereda de padres a hijos según un patrón autosómico recesivo, lo cual significa que los padres generalmente están sanos, no presentan el trastorno, pero son portadores y transmiten la anomalía a sus hijos. El niño debe recibir una copia del gen anómalo de cada progenitor para presentar la enfermedad.
La acidemia propiónica puede manifestarse según diferentes patrones clínicos, pero la forma más habitual es la severa neonatal, en la que aparecen los síntomas en la primera semana de vida y se manifiesta por vómitos, perdida de peso, síntomas neurológicos, perdida de tono muscular y convulsiones, todo ello puede llevar al coma. En los exámenes de laboratorio se detecta acidosis metabólica y elevación de NH4+ en sangre hiperamoniemia
Este trastono se incluye dentro del grupo de enfemedades llamadas errores congénitos del metabolismo y su frecuencia es variable según la región, en Estados Unidos se presenta un caso por cada 35000 nacimientos, mientras que en Arabia Saudi afecta a 1 de cada 3000 nacidos.
Se hereda de padres a hijos según un patrón autosómico recesivo, lo cual significa que los padres generalmente están sanos, no presentan el trastorno, pero son portadores y transmiten la anomalía a sus hijos. El niño debe recibir una copia del gen anómalo de cada progenitor para presentar la enfermedad.
La acidemia propiónica puede manifestarse según diferentes patrones clínicos, pero la forma más habitual es la severa neonatal, en la que aparecen los síntomas en la primera semana de vida y se manifiesta por vómitos, perdida de peso, síntomas neurológicos, perdida de tono muscular y convulsiones, todo ello puede llevar al coma. En los exámenes de laboratorio se detecta acidosis metabólica y elevación de NH4+ en sangre hiperamoniemia
Abetalipoproteinemia
Abetalipoproteinemia
La abetalipoproteinemia o Síndrome de Bassen-Kornzweig, es una rara enfermedad que afecta al tracto digestivo, cuya principal característica es la incapacidad que tiene el organismo de absorber adecuadamente los componentes grasos del alimento a través del intestino, lo que tiene como consecuencia la producción de heces grasosas (esteatorrea), deficiencia en el desarrollo infantil y problemas en los nervios.Esta enfermedad es de origen genético, probándose la existencia de una mutación en un gen en los pacientes: proteína de transferencia de triglicérido microsómico (MTP). Esta enfermedad afecta a ambos sexos, sin embargo es más frecuente en los hombres, llegando estos a representar el 70% de los casos
El síndrome de Aase o síndrome de Aase-Smith
A - Z
Es una enfermedad rara, hereditaria caracterizada por anemia asociada a malformaciones articulares y esqueléticas. Se trata de una enfermedad autosómica dominante. La alteración genética es desconocida. La anemia es causada por una displasia de médula ósea. Recibe su nombre en honor de los pediatras estadounidenses Jon Morton Aase y David Weyhe Smith.
Síntomas

- Alteración en crecimiento
- Palidez cutánea
- Retraso en el cierre de fontanelas
- Hombros estrechos
- Triple articulación del pulgar, pequeños o ausentes nudillos, disminución de pliegues cutáneos en articulaciones de los dedos
- Imposibilidad de extensión articular desde nacimiento (contractura congénita)
ENFERMEDADES RARAS
Las enfermedades raras, minoritarias o huérfanas, incluidas las de origen genético, son aquellas enfermedades que afectan a un pequeño número absoluto de personas o a una proporción reducida de la población.
Suscribirse a:
Entradas (Atom)